- What is intOGen?
- How does intOGen work?
- What is new in this release?
- Why do some driver genes from the previous release not appear in the latest release?
- How did you gather all these samples?
- How do you pre-process the input somatic point mutations?
- Which methods for cancer driver gene identification are used?
- How do you combine the output of the seven methods?
- How do you quantify the credibility of each method for a given cohort?
- What is the performance of the combination?
- Do you post-process the raw output of the combination to produce the final compendium of driver genes?
- What are the mutational pattern features?
- How did you map the publicly available cohorts into cancer types?
- Which genomic transcript is chosen for each gene?
- Are you planning to include new datasets?
- Are you planning to include new methods?
- What is the meaning of in silico saturation mutagenesis?
- How do I cite intOGen?
- Which is the intOGen License?
- What did it take to develop intOGen?
- Who contributed to intOGen?
- Can I run the intOGen pipeline locally?
- Is the code of the intOGen pipeline open source?
- Where do I download the full compendium of driver genes and their mutational features?
- Can I download the raw list of driver genes per cohort?
- What human genome assembly is used?
- Can I run locally in GRCh37?
- Can I reproduce your post-processing?
- Why driver genes do not have mutational features when a tumor type is not selected?
- Can I access to previous releases of intOGen?
- Can I provide feedback?
- Why does this site use cookies and what for?
What is intOGen?
IntOGen is a framework for automatic and comprehensive knowledge extraction based on mutational data from sequenced tumor samples from patients. The framework identifies cancer genes and pinpoints their putative mechanism of action across tumor types.
How does intOGen work?
Given a dataset of somatic point mutations from a cohort of tumor samples, intOGen first pre-processes the input mutations, next it runs seven different methods for cancer driver gene identification and, finally, it combines the output of these methods to produce a compendium of driver genes and a repository of the mutational features that can be used to explain their mechanisms of action.
What is new in this release?
We have upgraded the driver identification pipeline (https://intogen-plus.readthedocs.io/en/v2024/) and we have updated Hartwig Medical Foundation dataset to version 2023. More information is provided in the Release notes.
Why do some driver genes from the previous release not appear in the latest release?
There are 633 driver genes in the latest release; among these, 50 are new and 36 included in the previous release are no longer identified as drivers. This is due to several reasons:
- Different mutation annotations:
- Refined filter for mutations selected as input for the methods.
- New canonical transcript selection (MANE transcripts), where annotation of mutations per gene may have changed with respect to the previous release
- Different bona fide cancer gene / artifacts lists:
- CGC genes: The step of combination in the pipeline depends on the list of bona fide cancer genes obtained from CGC.
- The final ranking q-value list:
- Everytime we run Intogen, the final ranking of q-values may change, due to heuristic calculation of the background model in different methods. This means that genes that are usually found close to the driver threshold may have driver or passenger status across runs. In this release, we included a seed option for 4 methods (those that allowed it), to reduce the variability across runs. This problem is still not solved, as there are still 3 methods where the background calculation cannot be fixed with a seed.
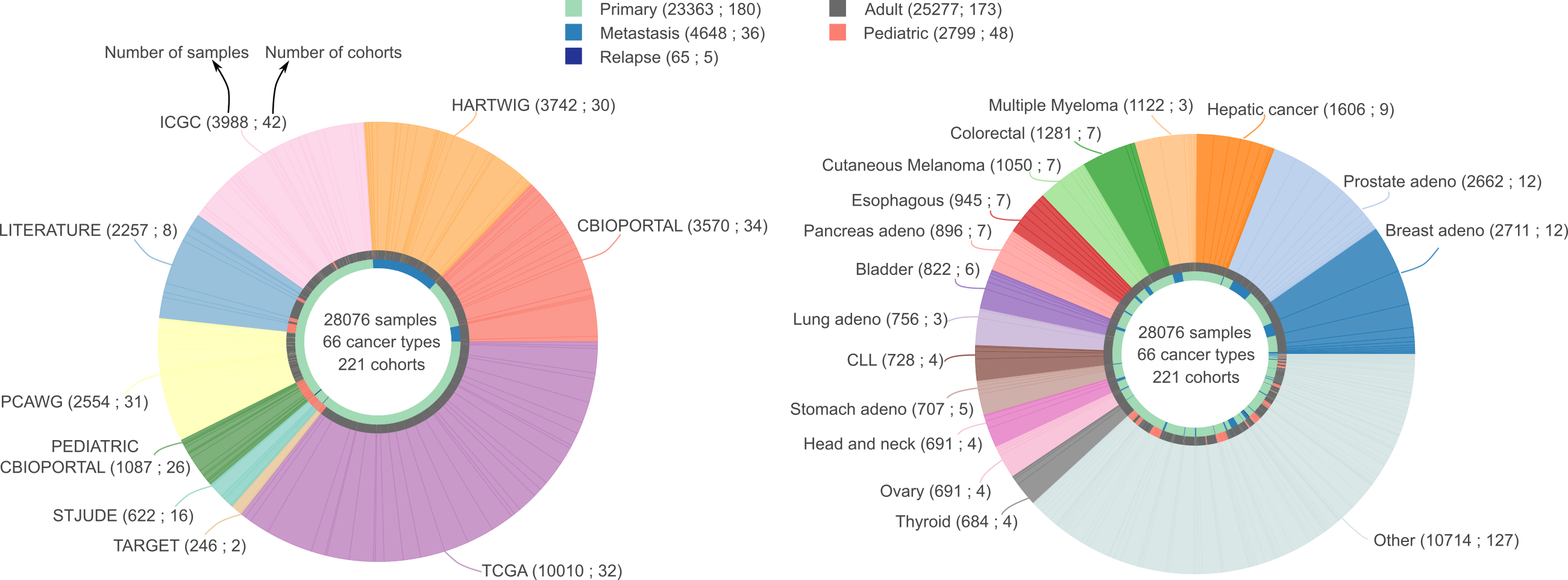
How did you gather all these samples?
We have manually downloaded and annotated tumor samples from different sources. Specifically, we have used cBioPortal, pediatric cBioPortal, ICGC, TCGA, PCAWG, Hartwig Medical Foundation, TARGET, St. Jude, CPTAC , CGCI and literature gathered sequencing projects projects. For a full list of cohorts included in this release, see the cohorts table in the Downloads tab. For further information about filtering and annotation please check our documentation.
Studies like ours would not be possible without the generosity of patients that decide to share their samples for research studies. They all deserve a thank you. We also thank the clinicians and researchers involved in obtaining the data for making it available and finally usable for others. We hope to make a convincing case that accessible data is necessary to enable and accelerate progress in science.
How do you pre-process the input somatic point mutations?
Given the heterogeneous nature of the multiple datasets analyzed in the current release of intOGen (resulting from e.g. differences in the genome aligners, calling algorithms, sequencing coverage, sequencing strategy), we have implemented a pre-processing strategy aiming at reducing biases induced by non-homogeneous input data. We removed hypermutated samples, dubious somatic variant calls, multiple samples from the same donor, datasets with pre-filtered synonymous mutations and mutations in regions with low mappability. For further information about the pre-processing steps please read our documentation.
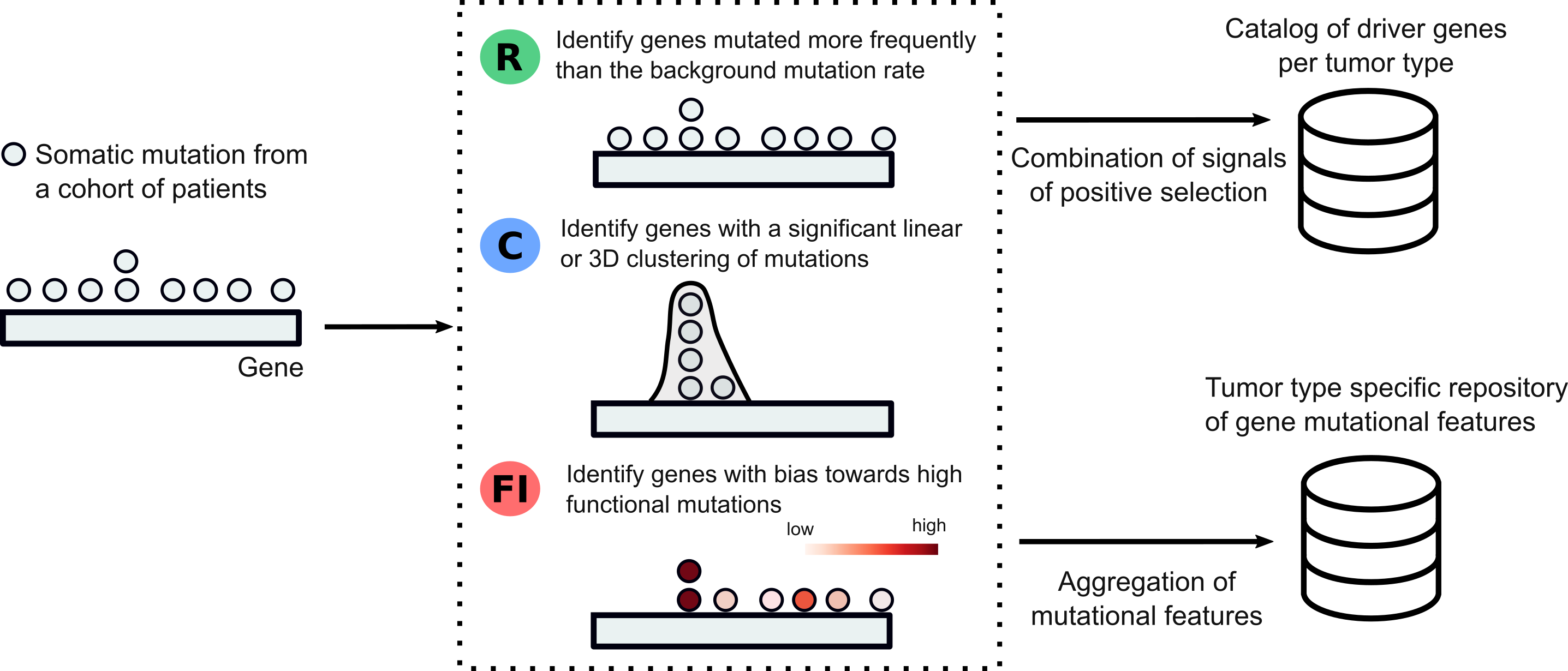
Which methods for cancer driver gene identification are used?
The current version of the intOGen pipeline uses seven methods to identify cancer driver genes from somatic point mutations. We used three methods, dNdScv, CBaSE and MutPanning which test for mutation count bias in genes while correcting for genomic covariates, mutational processes and coding consequence type; three methods that test for significant clustering of mutations in the protein sequence (OncodriveCLUSTL), protein structure (HotMAPS), and protein functional domains (smRegions); and one method that tests for functional impact bias of the observed mutations (OncodriveFML).
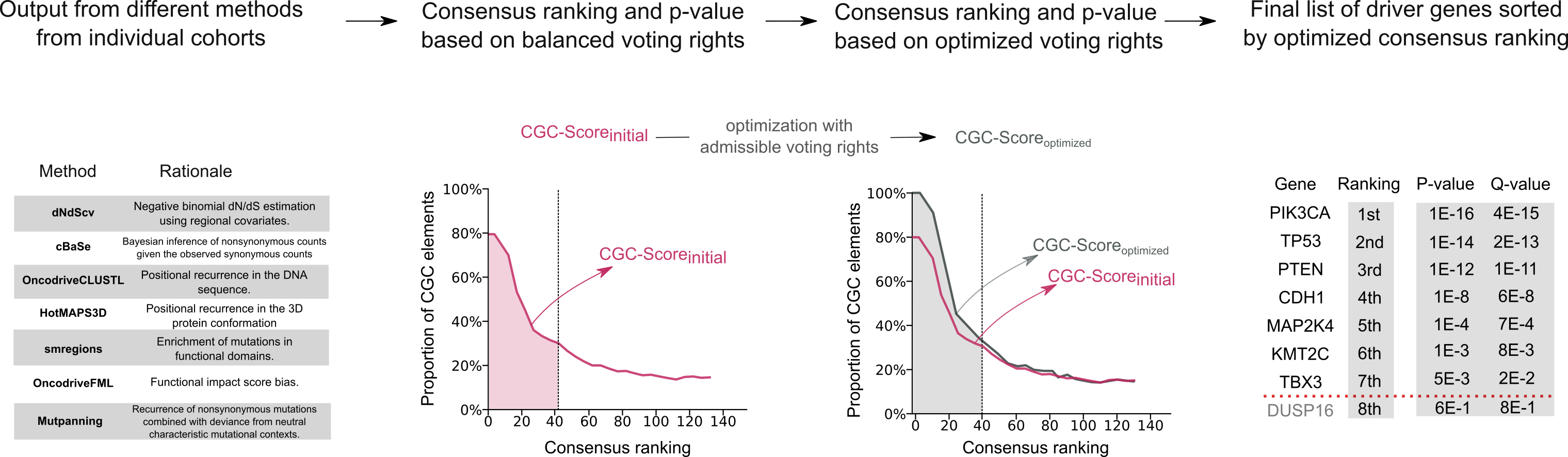
How do you combine the output of the seven methods?
Our approach works independently for each cohort: to create a consensus list of driver genes for each cohort, we first determine how credible each method is when applied to this specific cohort, on the basis of how many bona fide cancer genes reported in the COSMIC Cancer Gene Census database (CGC) are highly ranked according to the method. Once the credibility of each method has been quantified, we use a weighted method for combining the p-values that each method produces for each candidate gene. This combination takes the methods credibility into account. Based on the combined p-values, we conduct FDR correction to conclude a ranking of candidate driver genes alongside q-values.
How do you quantify the credibility of each method for a given cohort?
The relative credibility for each method is based on the ability of the method to give precedence to well-known genes already collected in the CGC catalogue of driver genes. As each method yields a ranking of driver genes, these lists can be combined using a voting system - based on the so-called Schulze’s method. Instead of conducting balanced voting, we tune the voting rights of the methods so that we maximize the enrichment of CGC genes at the top positions of the consensus list upon voting. In order to prevent degenerate solutions, we impose some constraints so that every method contributes with a minimum share. We also limit the combined share of coalitions of methods based on similar signals of positive selection. The solution voting rights are deemed the relative credibility for each method.
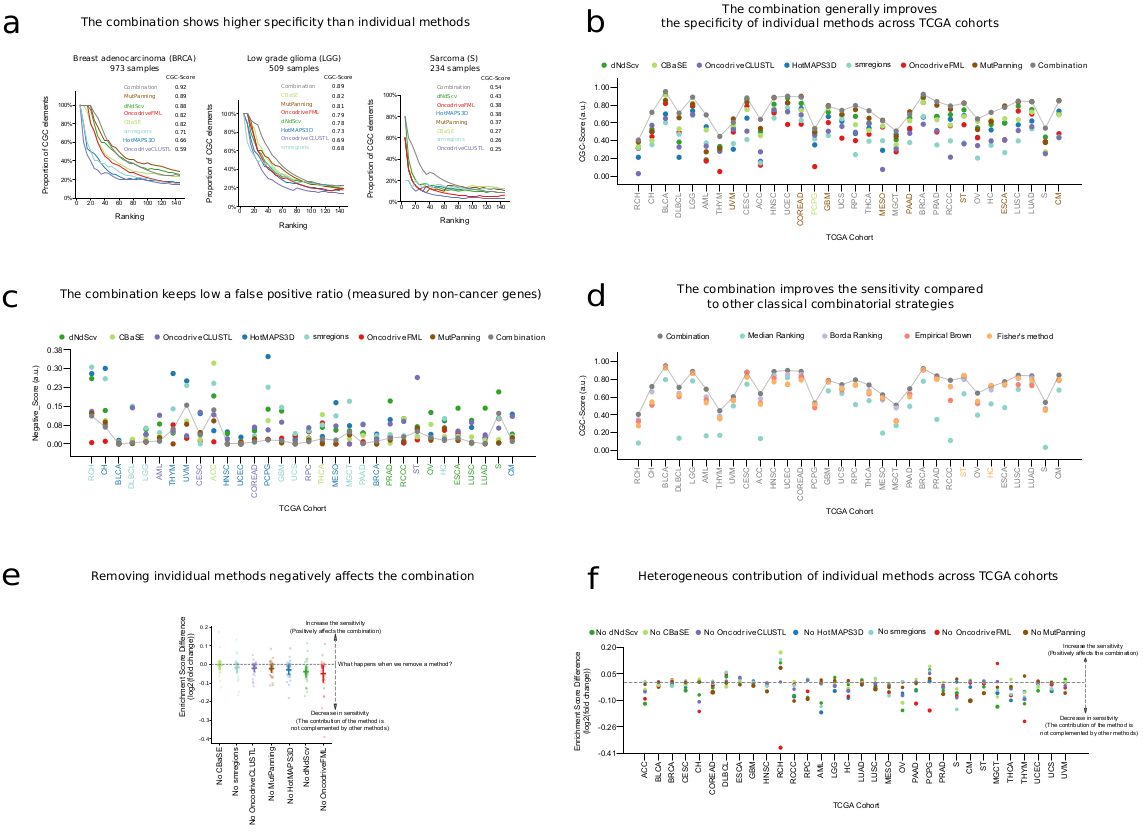
What is the performance of the combination?
Disclaimer: This analyses was performed on IntOGen v2020. When compared to individual methods, our combination method achieves highest enrichment of CGC genes among top ranked genes (CGC enrichment score; see panels A and B). When compared to commonly used alternative combination methods based on the same individual outcomes, our proposed combination tended to yield highest CGC enrichment score (panel D). We also checked the enrichment of known false discovery artifacts (e.g. long, late-replicating, structural and/or inactive genes such as TTN or olfactory receptors) and found that our combination method tended to yield lower false positives than individual methods (panel C). Finally, we also assessed the effect on the CGC enrichment score upon leaving each method out before combining, one method at a time: with few exceptions, the effect of leaving any method out was detrimental to the CGC enrichment score, meaning that in general all the methods are effectively required to reach the optimal consensus attained (panel E).
Do you post-process the raw output of the combination to produce the final compendium of driver genes?
The intOGen pipeline outputs a ranked list of driver genes per input cohort. We aimed to create a comprehensive compendium of driver genes per tumor type from all the cohorts included in this version. Then, we performed a filtering on automatically generated driver gene lists per cohort. This filtering is intended to lessen artifacts from the cohort-specific driver lists (e.g., due to errors in calling algorithms, errors introduced by positive selection methods, local hypermutations effects, undocumented filtering of mutations). Further details of the post-processing step can be found in intOGen documentation (or here for a .pdf version).
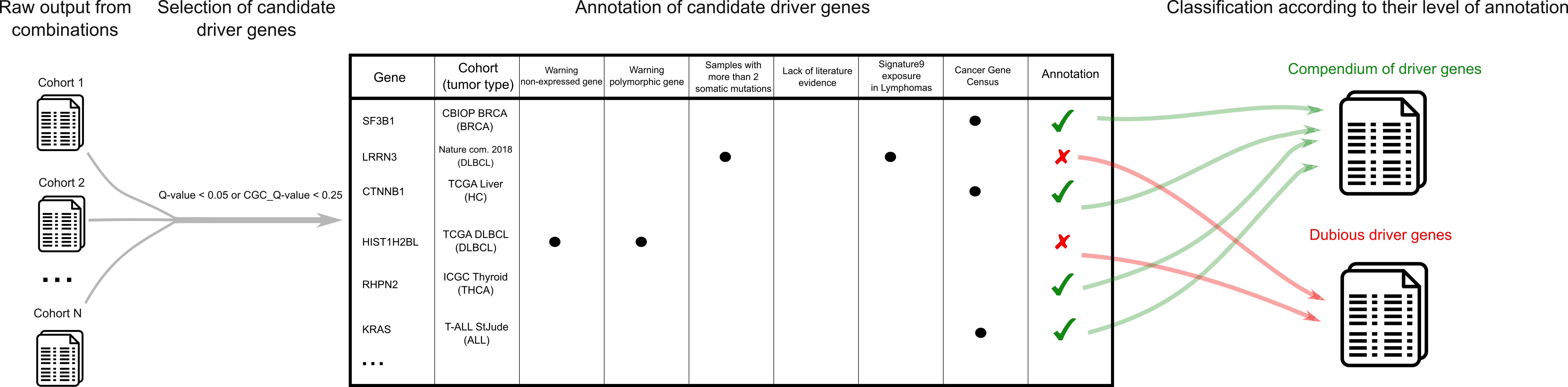
What are the mutational pattern features?
The abnormal mutational patterns that underpin the detection of positive selection in the driver genes across tumors also inform on their possible role in tumorigenesis. In addition to the compendium of cancer driver genes, the pipeline also generates a database where these mutational patterns are annotated as features. Some of these features are computed by the driver discovery methods employed in the drivers identification pipeline. Others are retrieved from public databases.
Specifically, we obtain the clusters of mutations along the sequence of proteins or within their 3D structure via OncodriveCLUSTL and HotMaps, respectively. We also retrieve the enrichment of mutations in a driver gene for protein Pfam domains through the smRegions methods. Finally, the excess of mutations derived from dNdS analysis for the different coding consequence types on the protein provides information on the mode of action of the driver gene in tumorigenesis.
How did you map the publicly available cohorts into cancer types?
We manually annotated the tumor type associated for each of the 266 input cohorts to a cancer type. Further information about mapping of each input cohort can be downloaded from Downloads.
Which genomic transcript is chosen for each gene?
We use the MANE transcript (v 1.2) defined by ENSEMBL and RefSeq as the reference transcript for each gene in our analysis. The current release uses VEP.111 from human GRCh38 genome assembly.
Are you planning to include new datasets?
Yes, we plan to incorporate new cohorts when new cancer datasets become publicly available. We will update the compendium of driver genes and mutational features accordingly. We are particularly interested in datasets of underrepresented cancer types, metastatic cohorts and pediatric tumors. Please email us to bbglab@irbbarcelona.org if you have suggestions about datasets that are not present and could be included.
Are you planning to include new methods?
We are always interested in updating and improving our driver discovery pipeline. Please email us to bbglab@irbbarcelona.org if you have suggestions about driver discovery methods that are not included and could be of potential interest.
What is the meaning of in silico saturation mutagenesis?
By in silico saturation mutagenesis of cancer genes we refer to the computational analysis whereby, by using mutational features, we can score and classify all possible single base substitutions in the canonical transcriptome of cancer genes by their potential to be involved in tumorigenesis. For this purpose, we developed a computational method so-called boostDM. The approach taken by boostDM is based on the analysis of observed mutations in sequenced tumors and their site-by-site annotation with relevant features. We leverage a series of mutational features identified across the sequenced tumors of IntOGen to build an ensemble of gene and tumor type-specific machine learning models intended to capture the making of their driver mutations. These models are trained on observed and randomly generated mutations across genes and tumors grouped by tumor type. The user can check the dedicated boostDM web app where the results for a collection of cancer genes and tumor types have been made publicly available. For more details about the methodology, validation and downstream analyses conducted with this tool, the user can also check the manuscript.
How do I cite intOGen?
If you find this resource useful please cite “Francisco Martínez-Jiménez, Ferran Muiños, Inés Sentís, Jordi Deu-Pons, Iker Reyes-Salazar, Claudia Arnedo-Pac, Loris Mularoni, Oriol Pich, Jose Bonet, Hanna Kranas, Abel Gonzalez-Perez & Nuria Lopez-Bigas A compendium of mutational cancer driver genes. Nature Reviews Cancer 2020; doi:10.1038/s41568-020-0290-x”. Also please link back to intOGen web if you use intOGen data.
Additionally, please consider citing the individual driver discovery methods used in the pipeline:
Which is the intOGen License?
All data released by intOGen aims to benefit the scientific community. The data released by intOGen is available free of restrictions under the Creative Commons Zero Public Domain Dedication. This means that you can use it for any purpose without legal need to give attribution. However, we kindly request that you actively cite and give attribution to our project, linking back to the relevant web page, wherever possible. Fair attribution supports future efforts and ensures correct legacy of the data.
The intOGen pipeline incorporates seven methods for cancer discovery. Hence, the source code distribution license needs to accommodate diverse agreements and licenses. The source code of the intOGen pipeline is provided under The GNU General Public License v3.0 .
What did it take to develop intOGen?
IntOGen has come about as a result of many different tasks that took the effort of a multidisciplinary team of scientists and engineers in differents areas of expertise: 1) surveying the literature, testing, adjusting and configuring the individual driver discovery methods; 2) conceptualizing, implementing and testing the driver discovery combination strategy; 3) conducting benchmarking analyses; 4) collecting and curating the publicly available datasets that intOGen relies on; 5) conceptualizing and implementing suitable pre-processing and post-processing; 6) implementing the intOGen workflow; 7) implementing the intOGen website; 8) preparing the figures and documentation; 9) maintaining the HPC infrastructure to carry out all the tests and analyses; 10) following-up, putting ideas together and discussing the most suitable features and steps forward; 11) leading and coordinating the team’s work.
Who contributed to intOGen?
IntOGen is a team effort from the Biomedical Genomics lab (https://bbglab.irbbarcelona.org/) at the Institute for Research in Biomedicine (IRB Barcelona).
Who has contributed to this release of IntOGen?
Mònica Sánchez Guixé prepared the Hartwig Medical Foundation data with the new tumor type annotations according to the DOID identification. Federica Brando and Miguel L Grau implemented the changes to the IntOGen pipeline described in the Release Notes, with Ferran Muiños participating in the discussions for the implementation related to the BoostDM connection. Federica Brando implemented the IntOGen website for this release. Ferran Muiños, Santiago Demajo and Stefano Pellegrini participated in discussions that helped to conceptualize and develop specific features of the pipeline. Joan Enric Ramis participated in the evaluation of the current release results. Mònica Sánchez Guixé, Federica Brando, Abel Gonzalez-Perez and Nuria Lopez-Bigas supervised and coordinated all the steps of the development of the current release of intOGen.
Who has contributed to the development of IntOGen over the years?
The methods currently used in IntOGen were tested and adjusted by Francisco Martínez-Jiménez, Ferran Muiños, Loris Mularoni and Claudia Arnedo-Pac. The main conceptualization of the combination strategy was carried out by Ferran Muiños and Francisco Martínez-Jiménez, who also prepared the figures and documentation in the IntOGen web. This strategy was benchmarked by Ferran Muiños, Francisco Martínez-Jiménez and Loris Mularoni. The cohorts included up to the previous IntOGen release were collected and curated by Mònica Sánchez-Guixé, Ines Sentís and Francisco Martínez-Jiménez. The current IntOGen pipeline was implemented by Federica Brando, Loris Mularoni, Jordi Deu-Pons, Iker Reyes-Salazar and Francisco Martínez-Jiménez. The framework of the IntOGen website was implemented by Federica Brando, Jordi Deu-Pons and Iker Reyes-Salazar. The conceptualization of the pipeline and the development of specific features were supported by discussions with other bbglab members, including Oriol Pich, Claudia Arnedo-Pac, Jose Bonet and Hanna Kranas. The development of the current IntOGen pipeline and the past data release was coordinated and supervised by Francisco Martínez-Jiménez, Abel Gonzalez-Perez and Nuria Lopez-Bigas.
Can I run the intOGen pipeline locally?
Yes, we have implemented a Nextflow pipeline that can be run locally. Please, bear in mind that intOGen pipeline runs seven driver discovery methods and it requires substantial amount of resources. We provide a step-by-step explanation of how to install and run the intOGen pipeline in the following link.
Is the code of the intOGen pipeline open source?
Yes, it is. Our aim is that all the scientific community can access to the pipeline and the results from the analysis presented in the website. Hence, you can access the source code at https://github.com/bbglab/intogen-plus/tree/v2024.
Where do I download the full compendium of driver genes and their mutational features?
In the Downloads tab you will have access to the latest (and previous) releases of the compendium of driver genes and mutational features.
Can I download the raw list of driver genes per cohort?
Yes, the raw output of the pipeline can be downloaded from the Downloads tab. Even though the combination improves the sensitivity and specificity compared to the output of the individual driver discovery methods, there are still some technical and biological caveats that may confound driver discovery in a systematic, unbiased way, thereby leading to false discovery of driver gene candidates. These artifacts include, e.g., low-quality mutation calling, local hypermutation events, not accounted for variability of the background mutation rate.
What human genome assembly is used?
All the methods are implemented to run in GRCh38, but the pipeline accepts output in GRCh37 and GRCh38.
Can I run locally in GRCh37?
Yes, the pipeline accepts somatic mutations in GRCh37 coordinates. However, all the pipeline works using GRCh38 coordinates. If GRCh37 assembly is selected the pipeline performs a liftover to GRCh38 coordinates.
Can I reproduce your post-processing?
Yes, the source code to post-process intOGen pipeline’s output is available at https://github.com/bbglab/intogen-plus/tree/v2024. Please check also the documentation of intOGen where all the post-processing steps are fully described.
Why driver genes do not have mutational features when a tumor type is not selected?
Genes have different mutational landscapes across different tissues and tumor types. Therefore, mutational features resulting from their tissue-specific distribution can only be found when a tumor type is selected.
Can I access to previous releases of intOGen?
Yes, if you want to download to datasets from previous releases from intOGen please visit the Downloads tab. Alternatively, if you are interested in browsing the previous release of the intOGen website, please go to https://www.intogen.org/legacy.
Can I provide feedback?
We are really interested in improving the quality and usability of intOGen. Therefore, if you have any technical concern please use the bitbucket tracker issue system at https://bitbucket.org/intogen/intogen-plus/issues?status=new&status=open. For any other issue, suggestion or proposal please contact with bbglab@irbbarcelona.org.
Why does this site use cookies and what for?
We are using Google Analytics cookies to track usage of our site. IntOGen is a publicly-funded project and these metrics are important to keep support for this project.