IntOGen plus - Release notes
What's new:
Release v2024.09.20
In this release we have re-organised the distribution of the Hartwig samples into cohorts using the DOID annotation. We have also adopted MANE (Matched Annotation from NCBI and EMBL-EBI) transcripts as the base to map mutations to protein coordinates. In addition to that, we updated the CGC annotation to the latest CGC v99, and carried out other small fixes. The current results show a modest increase in the number of driver genes (N=633 vs 619). The vast majority (N=583) of driver genes in the previous release are still in the Compendium, which also includes previously unidentified drivers. Conversely, N=36 genes in the previous release –mostly identified in a single cohort– are now absent from the Compendium. Most of these differences are directly caused by the change of reference transcript to the MANE, which resulted in the inclusion or exclusion of mutations overlapping the protein coding sequence. We show some examples of both cases.
Know more..
Here we describe in detail all changes carried out in this release: the new Hartwig cohorts organization, the updates on the pipeline, and how these changes affect the discovery of driver genes.
Update Hartwig dataset to v2023
We have updated the Hartwig dataset with the data release of September 2023. We have changed the tumor type annotation for each sample in Hartwig: instead of a manual annotation using the primary tumor type descriptions, we are now using the DOID identification to annotate the corresponding Oncotree code. In the case of tumor types with less than 20 samples, we re-assigned the tumor type to a parent node of the Oncotree, up until we can group at least 20 samples. This tumor type re-annotation of the entire Hartwig dataset modifies the current cohorts and new cohorts appear.

| New Cohorts | Lost Cohorts | ||||||
|---|---|---|---|---|---|---|---|
| Cohort name | Tumor type | Cohort name | Tumor type | ||||
| HARTWIG_WGS_BLADDER_2023 | Bladder/Urinary Tract | HARTWIG_WGS_BCC_2023 | Basal Cell Carcinoma | ||||
| HARTWIG_WGS_CEAD_2023 | Cervical Adenocarcinoma | HARTWIG_WGS_CESC_2023 | Cervical Squamous Cell Carcinoma | ||||
| HARTWIG_WGS_COAD_2023 | Colon Adenocarcinoma | HARTWIG_WGS_CSCC_2023 | Cutaneous Squamous Cell Carcinoma | ||||
| HARTWIG_WGS_EGC_2023 | Esophagogastric Adenocarcinoma | HARTWIG_WGS_GBM_2023 | Glioblastoma Multiforme | ||||
| HARTWIG_WGS_GBC_2023 | Gallbladder Cancer | HARTWIG_WGS_GINET_2023 | Gastrointestinal Neuroendocrine Tumors | ||||
| HARTWIG_WGS_GB_2023 | Glioblastoma | HARTWIG_WGS_LNET_2023 | Lung Neuroendocrine Tumor | ||||
| HARTWIG_WGS_GIST_2023 | Gastrointestinal Stromal Tumor | HARTWIG_WGS_LNM_2023 | Lymphoid Neoplasm | ||||
| HARTWIG_WGS_LIPO_2023 | Liposarcoma | HARTWIG_WGS_MCC_2023 | Merkel Cell Carcinoma | ||||
| HARTWIG_WGS_LMS_2023 | Leiomyosarcoma | HARTWIG_WGS_MEL_2023 | Melanoma | ||||
| HARTWIG_WGS_LUNG_2023 | Lung | HARTWIG_WGS_OS_2023 | Osteosarcoma | ||||
| HARTWIG_WGS_MT_2023 | Malignant Tumor | HARTWIG_WGS_PANET_2023 | Pancreatic Neuroendocrine Tumor | ||||
| HARTWIG_WGS_NETNOS_2023 | Neuroendocrine Tumor, NOS | HARTWIG_WGS_PRAD_2023 | Prostate Adenocarcinoma | ||||
| HARTWIG_WGS_PROSTATE_2023 | Prostate | HARTWIG_WGS_PSCC_2023 | Penile Squamous Cell Carcinoma | ||||
| HARTWIG_WGS_READ_2023 | Rectal Adenocarcinoma | HARTWIG_WGS_SBC_2023 | Small Bowel Cancer | ||||
| HARTWIG_WGS_SACA_2023 | Salivary Carcinoma | HARTWIG_WGS_STAD_2023 | Stomach Adenocarcinoma | ||||
| HARTWIG_WGS_SARCNOS_2023 | Sarcoma, NOS | HARTWIG_WGS_THYM_2023 | Thymoma | ||||
| HARTWIG_WGS_SIC_2023 | Small Intestinal Carcinoma | HARTWIG_WGS_VULVA_2023 | Vulva/Vagina | ||||
| HARTWIG_WGS_SKCM_2023 | Cutaneous Melanoma | ||||||
| HARTWIG_WGS_SKIN_2023 | Skin | ||||||
| HARTWIG_WGS_SOFT_TISSUE_2023 | Soft Tissue | ||||||
| HARTWIG_WGS_STOMACH_2023 | Esophagus/Stomach | ||||||
| HARTWIG_WGS_UTUC_2023 | Upper Tract Urothelial Carcinoma | ||||||
| Show more | |||||||
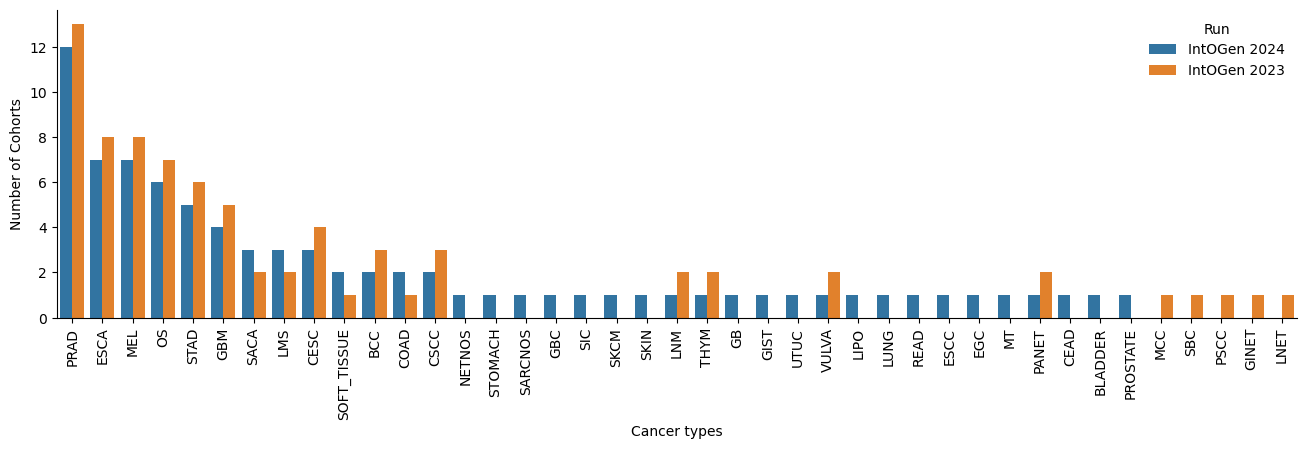
Updated pipeline
Several updates and important bugfixes have been introduced in the IntOGen pipeline:
- Ensembl VEP v111
- Use of MANE Transcripts
- Impact on datasets
- Impact on the pipeline
-
VepStep. The process now runs with –mane flag, that includes the column
MANE_SELECT. -
ProcessVEPoutput step.
The filtering out of the transcript is now based on the newly added
MANE_SELECTcolumn. The step only retains transcripts with a Refseq ID equivalent in theMANE_SELECTcolumn. Moreover we updated the list of accepted consequence type to match with the new VEP v111. - CGC v99
- cBaSE v1.1 update
- BoostDM connection (for further details in BoostDM updates please visit BoostDM release notes)
- DriversSaturation step
- ParseProfile step
- Renaming of Steps (Signature → ComputeProfile)
We have updated the Ensembl VEP from v101 to v111. This version differs substantially from the previous one used in IntOGen. Key differences are the redefinition of canonical transcripts and different annotations for some genes and mutations. Therefore, the number of mutations annotated to a gene may change from that in the previous release.
We have switched to using MANE transcript in our pipeline, to align with common standards used in the clinics.
Redefinition of the transcript impacts the regions used in methods like OncodriveFML, OncodriveCLUSTLand and smRegions, when computing the profile and when building coding sequence regions for dNdS.
We have updated the Cancer Gene Census gene list to v99, since COSMIC dropped support for releases prior v96.
Former version of cBaSE had a major bugfix when analyzing cohorts with low TMB that would call every gene a driver. This has been solved by updating from v1.0 to v1.1.
The regions are now defined as the CDS according to the MANE SELECT transcripts, including stop codons, plus 5bp segments flanking each CDS segment.
Profile output by ComputeProfile step is parsed in a 96-channel for every trinucleotide context, to ensure consistency and compatibility with BoostDM run.
We renamed the Signature step into a more self-explainable name: ComputeProfile, in order to avoid confusion and misunderstanding.
New results
We have obtained a new set of 633 driver genes. The list of driver genes may change with respect to the previous release of the Compendium due to several reasons:
- The change of canonical transcript for the MANE select transcript.
- The update of COSMIC CGC to v99
- The final ranking q-value list. As explained in the previous release, the final ranking of q-values may change due to heuristic calculation of the background model in different methods.
| Intogen run | Total number of mutations | Total number of samples | Number of cancer types | Number of cohorts | Number of drivers |
|---|---|---|---|---|---|
| Release 2024 | 257,898,749 | 33,218 | 87 | 271 | 633 |
| Release 2023 | 252,486,809 | 33,019 | 73 | 266 | 618 |
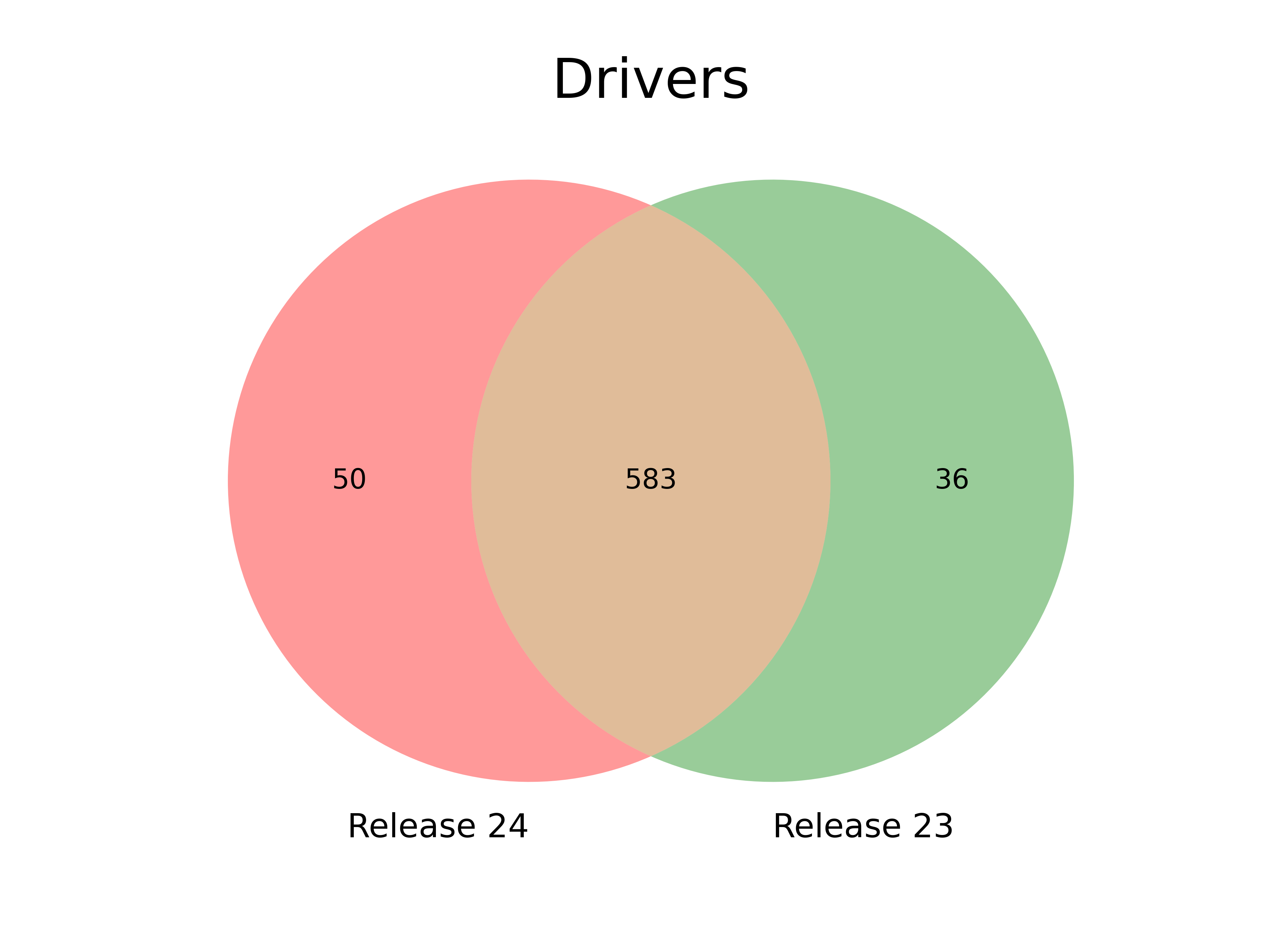
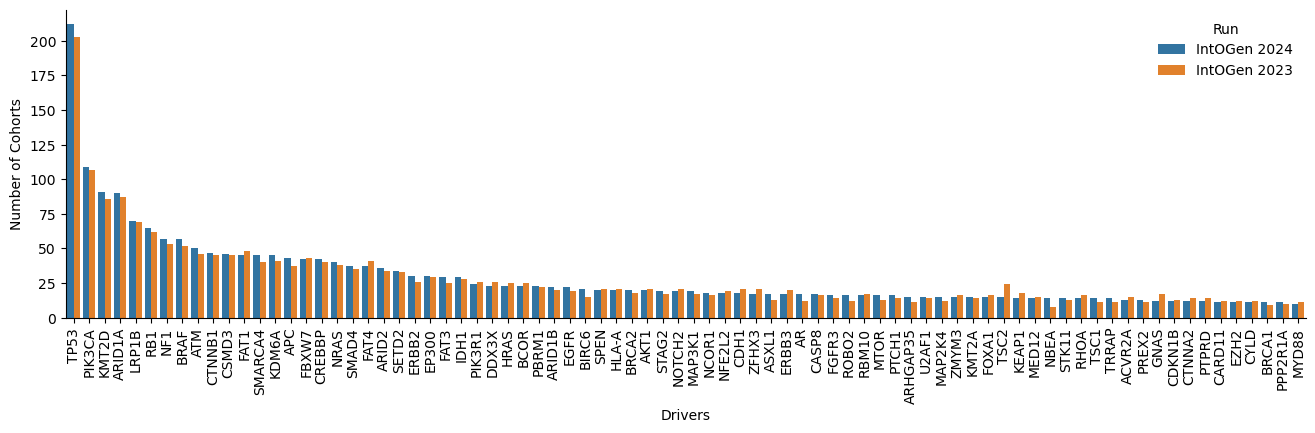
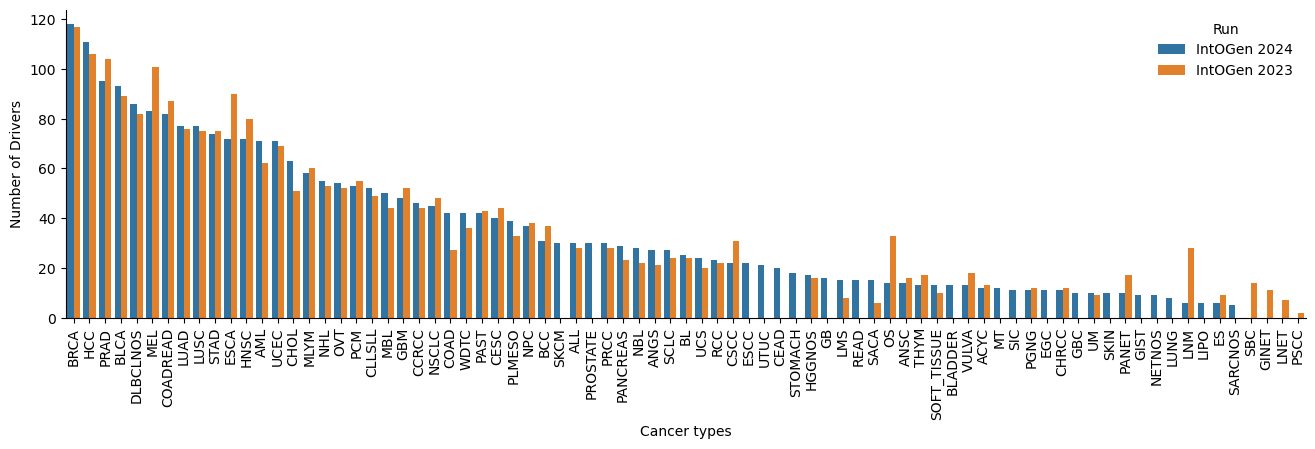
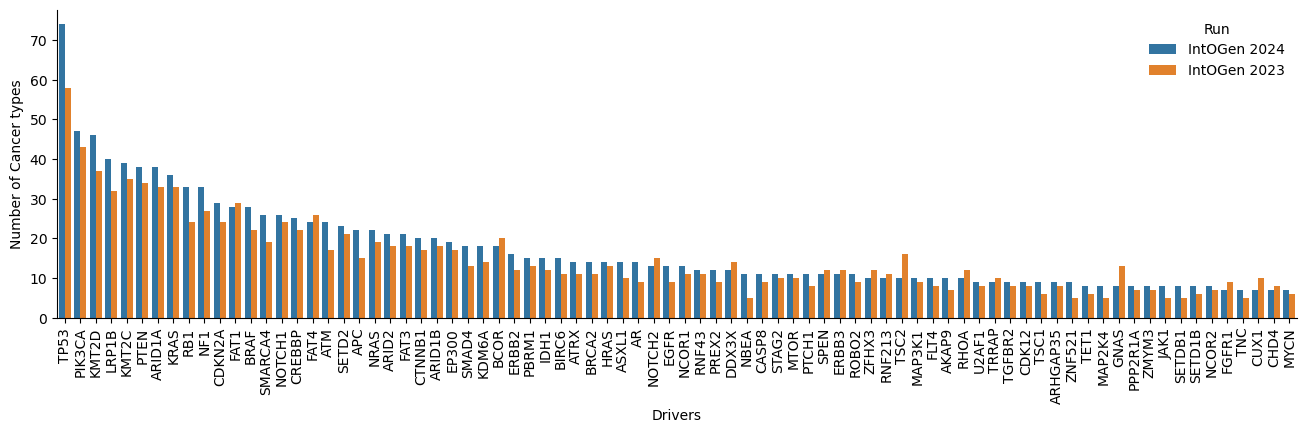
After further inspection on the new driver genes and the genes no longer called drivers, we observe that an important number (12 new drivers) change the status in the CGC database and also present a different canonical transcript (16 new drivers and 13 lost drivers). Below are some examples (Table 4 and 5, Figure 5 to 7).
| Different Transcript | Different ranking in CGC | Number of New Drivers | Number of Lost Drivers |
|---|---|---|---|
| True | True | 4 | / |
| False | 12 | 13 | |
| False | True | 8 | / |
| False | 39 | 30 |
| Gene | Transcript (v111) | Transcript (v101) | Different Transcript | Cohort name | Gene in CGC v95 | Gene in CGC v99 | Different CGC ranking |
|---|---|---|---|---|---|---|---|
| NACA | ENST00000454682 | ENST00000550952 | True | PCAWG_WGS_SKIN_MELANOMA, TCGA_WXS_MEL, HARTWIG_WGS_BLCA_2023, CBIOP_WXS_BCC_UNIGE_2016_UNTREAT | True | True | False |
| PRKD1 | ENST00000331968 | ENST00000415220 | True | CBIOP_WXS_PRAD_MSKCC_DCFI_2018_PRY, HARTWIG_WGS_COADREAD_2023, PCAWG_WGS_BILIARY_ADENOCA | True | False | True |
| NTRK2 | ENST00000277120 | ENST00000277120 | False | PEDCBIOP_WXS_MBL_SHH_PRY, ICGC_WXS_CLLSLL_CLLE_ES_2019, CBIOP_WXS_ANGS_TREATED_2020 | True | False | True |
| AFF3 | ENST00000672756 | ENST00000409579 | True | CBIOP_WXS_LUAD_VALLEN_2018, ICGC_WXS_WDTC_THCA_SA_2019 | True | True | False |
| CTNNA1 | ENST00000302763 | ENST00000302763 | False | ICGC_WXS_COADREAD_COCA_CN_CASPOINT_2019, HARTWIG_WGS_CEAD_2023 | True | False | True |
| RAD50 | ENST00000378823 | ENST00000378823 | False | CPTAC_WXS_GBM_2020 | True | False | True |
| NCOA1 | ENST00000348332 | ENST00000406961 | True | CPTAC_WXS_PAAD_2021 | True | True | False |
| RAP1B | ENST00000250559 | ENST00000250559 | False | HARTWIG_WGS_BLCA_2023 | True | False | True |
| TMPRSS2 | ENST00000332149 | ENST00000398585 | True | OTHER_WGS_PRAD_CPGEA_2021 | True | True | False |
| CRLF2 | ENST00000400841 | ENST00000381566 | True | TCGA_WXS_LUAD | True | True | False |
| ERG | ENST00000288319 | ENST00000417133 | True | CBIOP_WXS_ANGS_TREATED_2020 | True | True | False |
| GSK3B | ENST00000264235 | ENST00000316626 | True | CBIOP_WGS_STAD_ONCOSG_2018 | True | False | True |
| HGF | ENST00000222390 | ENST00000222390 | False | CBIOP_WXS_SCLC_CLCGP | True | False | True |
| HIF1A | ENST00000337138 | ENST00000539097 | True | ICGC_WXS_WDTC_THCA_SA_2019 | True | True | False |
| ZCCHC8 | ENST00000633063 | ENST00000543897 | True | CBIOP_WXS_EGC_TMUCIH_2015 | True | True | False |
| Gene | Transcript (v111) | Transcript (v101) | Different Transcript | Cohort name | Gene in CGC v95 | Gene in CGC v99 | Different CGC ranking |
|---|---|---|---|---|---|---|---|
| ETNK1 | ENST00000266517 | ENST00000671733 | True | ICGC_WXS_NPC_ORCA_IN_2019, OTHER_WGS_MM_NATLEUK2018 | True | True | False |
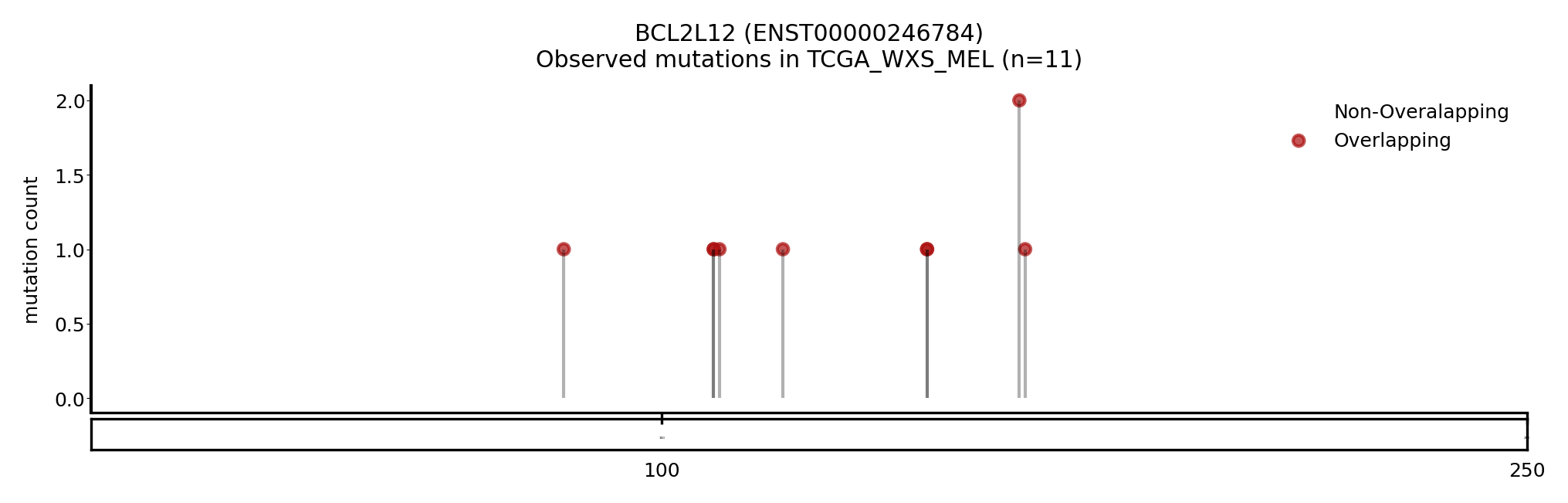
| BCL2L12 | ENST00000246784 | ENST00000616144 | True | CBIOP_WXS_CSCC_UCSF_2018, TCGA_WXS_MEL | True | True | False |
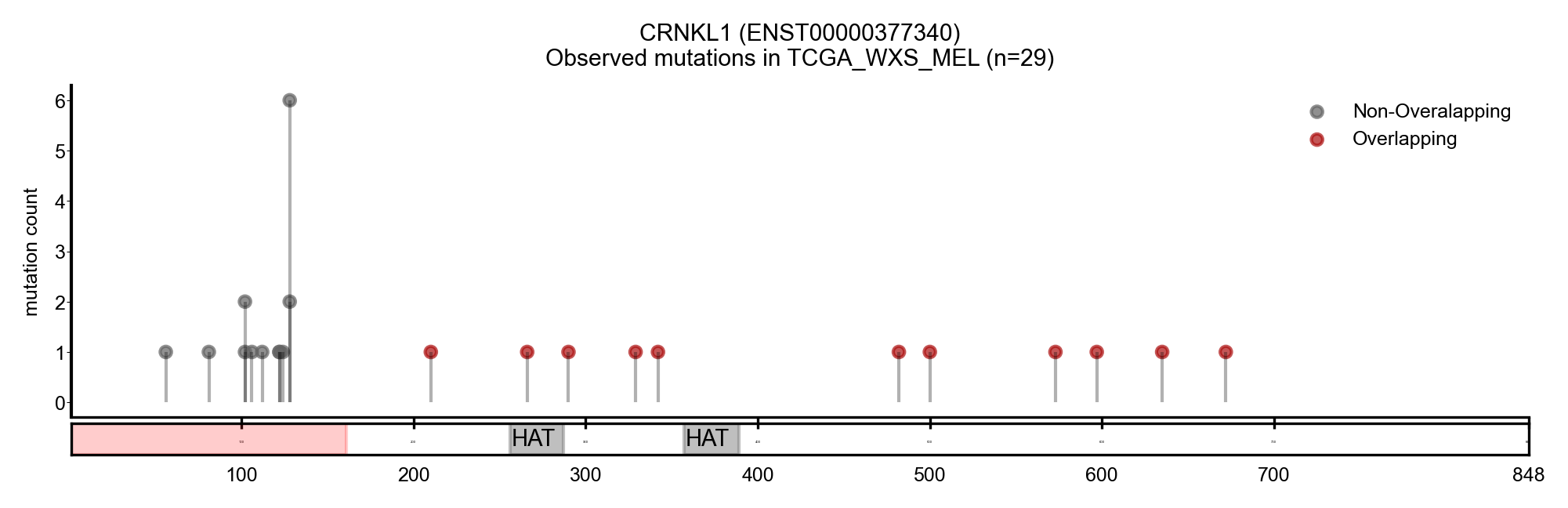
| CRNKL1 | ENST00000536226 | ENST00000377340 | True | CBIOP_WXS_CSCC_UCSF_2018, TCGA_WXS_MEL | True | True | False |
| TFEB | ENST00000373033 | ENST00000358871 | True | OTHER_WGS_PLMESO_MESOMICS_2023 | True | True | False |
| PDE4DIP | ENST00000695795 | ENST00000369356 | True | TCGA_WXS_OVT | True | True | False |
| SLC35E2A | NaN | ENST00000643905 | True | ICGC_WGS_LUSC_LUSC_KR_2019 | False | False | False |
| S100A7 | ENST00000368723 | ENST00000368722 | True | HARTWIG_WGS_OVT_2023 | True | True | False |
| PTPRU | ENST00000373779 | ENST00000345512 | True | TCGA_WXS_LUAD | False | False | False |
| FANCD2 | ENST00000675286 | ENST00000287647 | True | ICGC_WXS_COADREAD_COCA_CN_VARSCAN_2019 | True | True | False |
| CREM | ENST00000685392 | ENST00000345491 | True | CPTAC_WXS_GBM_2020 | False | False | False |
Here we show some examples of genes that change the driver status likely because they change the canonical transcript: a new driver, NACA, presented with a longer MANE transcript, containing a region with a cluster of mutations (Figure 5). On the other hand, BCL2L12 and CRNKL1 are two genes with a shorter transcript, losing a region that contains a cluster of mutations (Figure 6 and 7).




v2023.05.31
In this release we have increased the number of cohorts (and samples), we have made small updates to the IntOGen pipeline and we have updated key third-party dependencies. As a result, the list of driver genes has increased modestly (N=618). The vast majority of previous driver genes are still in this list, which also include previously unidentified drivers, while few others –mostly identified in a single cohort in the previous release– now fail to pass the thresholds set in the pipeline.
Know more..
Here we describe in detail all changes carried out in this release: the new cohorts and samples incorporated, the updates on the pipeline, and how these changes affect the discovery of driver genes.
More cohorts, more samples
In this new release we have incorporated 48 new cohorts to the analysis (Figure 1) representing 4943 new samples (Table 1 and Table 2).
Some previously included cohorts have increased the number of samples they contribute to the analysis. This is the case of cohorts sequenced by the Hartwig Medical Foundation (release downloaded on 21/10/2021) and ICGC (latest release from 2019 downloaded). After careful examination, we have excluded an ICGC cohort (LINC_JP_WGS_ICGC) which was completely included within the HC_PCAWG cohort (now re-named PCAWG_WGS_LIVER_HCC) (Figure 1).
New cohorts have been contributed by some already included data sources, such as TARGET, from which we obtained 2 new cohorts with Neuroblastoma (NB) and Osteosarcoma (OS) samples.
We also included datasets from new genomic data sources, such as CPTAC and CGCI.
Due to changes in the sample annotation of the TCGA_WXS_AML cohort, 95 more samples of this cohort were processed by the pipeline.
In order to standardize the cohort nomenclatures, cohort names have been updated, applying -in most cases- the following pattern:
{PROJECT}_{PLATFORM}_{ONCOTREE_CODE}_{OTHER_INFO}_{YEAR}
Examples: CPTAC_WXS_BRCA_2020, ICGC_WXS_AML_LAML_CN_2019, HARTWIG_WGS_NSCLC_2020
| Cancer type | Samples release 2023 | Samples release 2020 | Number of new samples | Increase (%) |
|---|---|---|---|---|
| Burkitt Lymphoma | 87 | 15 | 72 | 480.00% |
| Lung Neuroendocrine Tumor | 30 | 10 | 20 | 200.00% |
| Pleural Mesothelioma | 337 | 117 | 220 | 188.03% |
| Cutaneous Squamous Cell Carcinoma | 128 | 49 | 79 | 161.22% |
| Nasopharyngeal Carcinoma | 221 | 103 | 118 | 114.56% |
| Angiosarcoma | 71 | 34 | 37 | 108.82% |
| Lymphoid Neoplasm | 48 | 25 | 23 | 92.00% |
| Lung Adenocarcinoma | 1,215 | 756 | 459 | 60.71% |
| Renal Clear Cell Carcinoma | 948 | 620 | 328 | 52.90% |
| Osteosarcoma | 553 | 363 | 190 | 52.34% |
| Non-Small Cell Lung Cancer | 564 | 384 | 180 | 46.88% |
| Cervical Squamous Cell Carcinoma | 467 | 341 | 126 | 36.95% |
| Well-Differentiated Thyroid Cancer | 852 | 684 | 168 | 24.56% |
| Stomach Adenocarcinoma | 858 | 707 | 151 | 21.36% |
| Esophageal Adenocarcinoma | 1,139 | 945 | 194 | 20.53% |
| Lung Squamous Cell Carcinoma | 703 | 584 | 119 | 20.38% |
| Others | 24,798 | 22,339 | 2,459 | 11.01% |
| Total | 33,019 | 28,076 | 4,943 | 17.61% |
| Data source | Samples release 2023 | Samples release 2020 | Number of new samples |
|---|---|---|---|
| STJUDE | 622 | 622 | 0 |
| PCAWG | 2,554 | 2,554 | 0 |
| PEDCBIOP | 1,087 | 1,087 | 0 |
| TCGA | 10,105 | 10,010 | 95 |
| TARGET | 365 | 246 | 119 |
| CGCI | 192 | 0 | 192 |
| HARTWIG | 4,386 | 3,742 | 644 |
| OTHER | 2,915 | 2,257 | 658 |
| CBIOP | 4,566 | 3,570 | 996 |
| CPTAC | 1,076 | 0 | 1,076 |
| ICGC | 5,151 | 3,988 | 1,163 |
| Total | 33,019 | 28,076 | 4,943 |
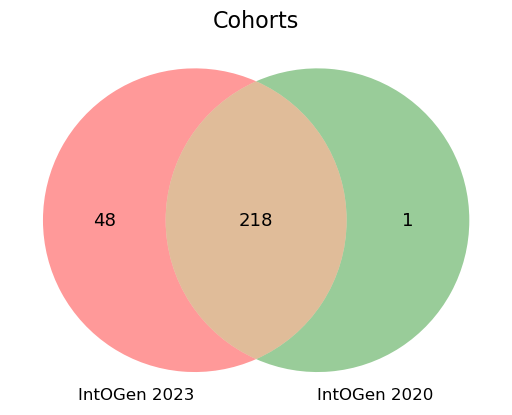
LINC_JP_WGS_ICGC).
Updated pipeline
Several updates have been introduced in the IntOGen pipeline:
- bgparsers replaced with OpenVariant:
- Seed to run OncodriveFML, OncodriveCLUSTL, smRegions and dNdScv added:
- Ensembl VEP updated from v92 to v101:
- MSKCC oncotree updated to version 2021:
NON_SOLIDnode added afterTISSUEand beforeMYELOIDandLYMPHOIDSOLIDnode added afterTISSUEand before the rest of tissuesALLnode added afterLNMand beforeTLLandBLL- CGC genes updated to v95:
- Hugo Symbols updated to the 20/01/2022 release:
- CancerMine database updated to 07/12/2021 release:
- CADD update:
- List of frequent artifacts updated:
- File with unfiltered drivers included:
- BoostDM connection prepared:
- DriversSaturation: all possible mutations for a given gene are generated and returned as independent annotated files per gene. [
{gene}.vep.gz] - filterMNVs: one file containing all positions detected as MNVs. [
mnvs.tsv.gz]
We have developed OpenVariant, a new python package to parse the input files. This new method annotates all SNVs and indels from the mutation data files and transforms the data in a standardized format, by reading a yaml file prepared by the user.
Importantly, the previous bgparsers tool had a bug in the annotation of indels, leading to some indels not being processed. This bug has been corrected in OpenVariant. As a consequence, there are, overall, more mutations annotated per sample.
We have added the use of a seed in these methods to reduce the variability in the calculation of the p-value of genes across IntOGen runs. Due to difficulties in the implementation, we could not add it in the remaining three methods run by the pipeline.
We updated the Ensembl VEP version to 101. This version may contain important gene annotation differences, and therefore the number of mutations annotated may change substantially from the previous release.
We have updated the oncotree from the latest version released by MSKCC (November 2021). As a result, the number of cancer type nodes has increased from 82 to 889.
We have applied some modifications to the MSKCC Oncotree:
We have updated the Cancer Gene Census gene list with the v95. This version distinguishes between Tier1 and Tier2 CGC genes. In the IntOGen pipeline, both tiers are considered as a unique list of CGC genes.
We have unified all gene SYMBOLs according to the latest version of the HUGO Gene Nomenclature Committee.
The CancerMine database, which gathers information on literature evidence for cancer genes, was updated.
CADD, a tool for scoring the deleteriousness of single nucleotide variants - as well as insertion/deletions variants in the human genome, was updated from version 1.4 to version 1.6.
In the previous release the pipeline was using a list of ‘Known Artifacts’ and a separated blacklist of genes, to rule out possible false positives in the post-processing. In this release, the blacklist genes are considered ‘Known Artifacts’, and, together with the ‘Warning artifacts’ (list of genes suspected to be artifacts) are all excluded in the post-processing step. Moreover, the olfactory receptors genes list from HORDE database used as a filter was updated to to Build #44c (30/July 2019).
In this new release we include a list of unfiltered drivers as one of the outputs of the IntOGen pipeline. This file annotates all the filters applied to the post-processing step: from the output of the combination to the final set of driver genes. [unfiltered_drivers.tsv]
We have included along with the IntOGen output, all the files needed to run BoostDM. Specifically, two steps in the pipeline were added:
New results
In the new release, several aspects have been improved: there are more cohorts and tumor type in which a gene is identified as driver; and more drivers and cohorts per tumor type (Figure 2).
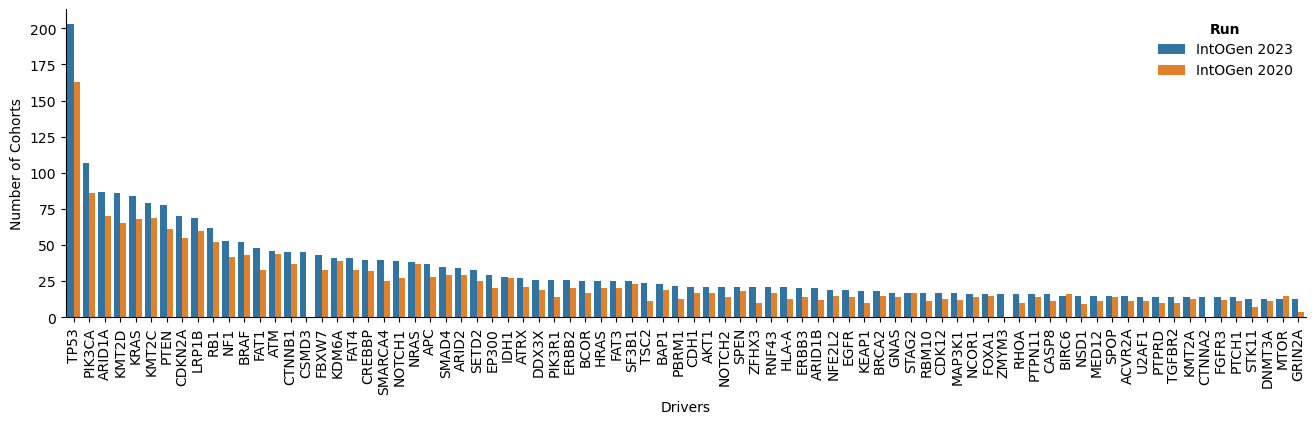
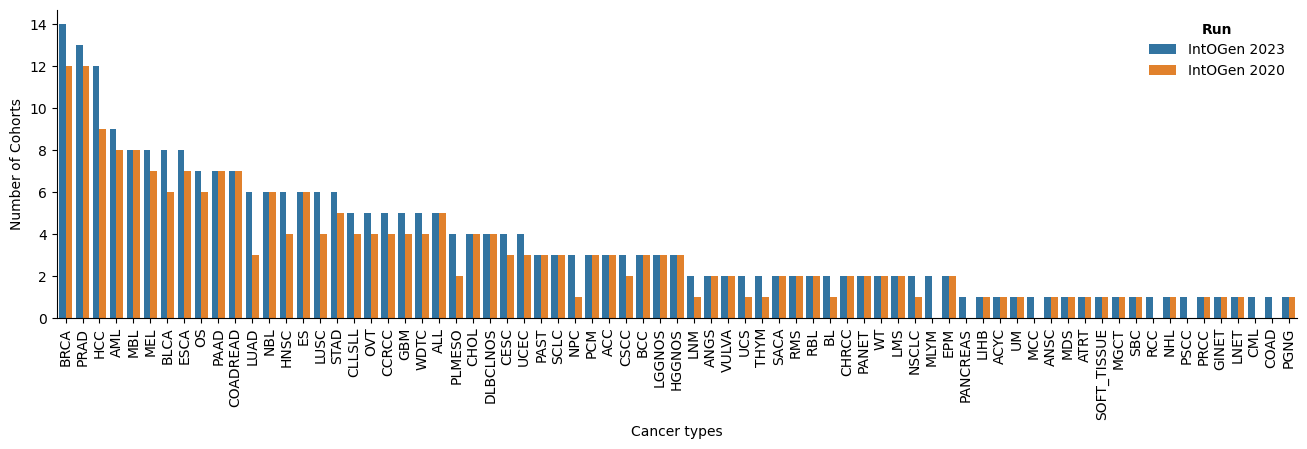
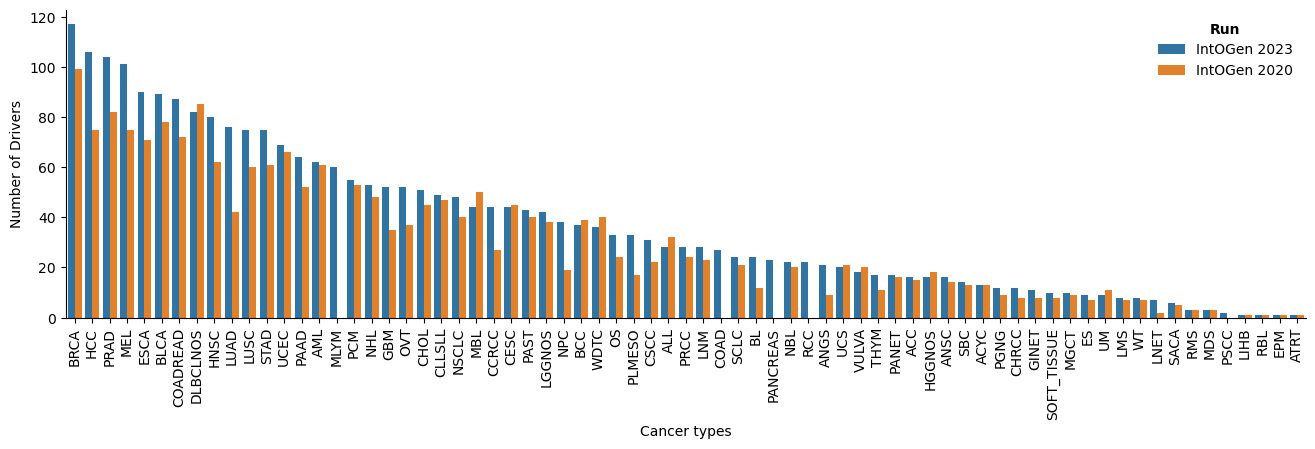
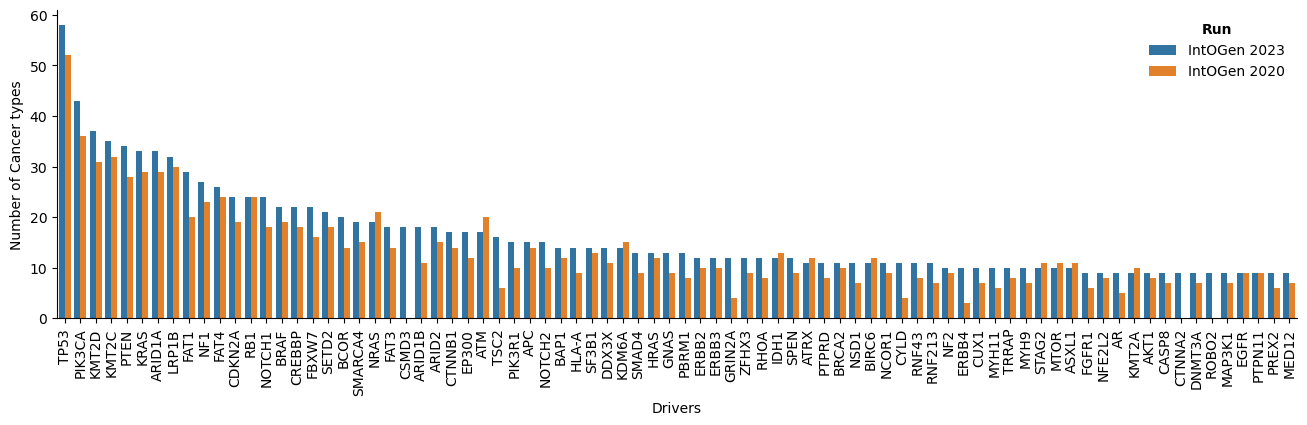
The new release contains a total of 618 driver genes, 50 more than the previous release. There are 7 new tumor types: two of them are from two cohorts with re-assigned tumor type
(from PAAD to PANCREAS on HARTWIG_PANCREAS and from RCCC to RCC on HARTWIG_KIDNEY_RENAL_CELL)
and five of them are new;
PSCC (Penil Squamous Cell Carcinoma, HARTWIG_WGS_PSCC_2020),
MCC (Merkel Cell Carcinoma, HARTWIG_WGS_MCC_2020),
COAD (Colon Adenocarcinoma, CPTAC_WXS_COAD_2020) and
MLYM (Malignant Lymphoma, ICGC_WGS_MLYM_MALY_DE_PED_2019)
| Intogen run | Total number of mutations | Total number of samples | Number of cancer types | Number of cohorts | Number of drivers |
|---|---|---|---|---|---|
| Release 2023 | 252,486,809 | 33,019 | 73 | 266 | 618 |
| Release 2020 | 203,003,747 | 28,076 | 66 | 221 | 568 |

Among these 618 driver genes, 133 that are new, while 82 included in the previous release are no longer identified as drivers (Figure 3). This is due to several reasons:
- Different mutation annotations:
- Increase in the number of processed indels (see above)
- New Ensembl gene annotation, where annotation of mutations per gene may have changed with respect to the previous release
- Different bona fide cancer gene / artifacts lists:
- CGC genes: The step of combination in the pipeline depends on the list of bona fide cancer genes obtained from CGC.
- CancerMine database: This database, that annotates literature-gathered information, is used to provide evidence of the involvement of genes in tumorigenesis.
- Lists of artifacts: in this release we filter out in the post-processing step all known artifacts and suspect/warning artifacts. The previous release was just filtering the known artifacts. These are accordingly annotated in the unfiltered drivers file.
- The final ranking q-value list:
- Implementation of a seed: Everytime we run Intogen, the final ranking of q-values may change, due to heuristic calculation of the background model in different methods. This means that genes that are usually found close to the driver threshold may have driver or passenger status across runs. In this release, we included a seed option for 4 methods (those that allowed it), to reduce the variability across runs. This problem is still not solved, as there are still 3 methods where the background calculation cannot be fixed with a seed.
We provide a table summarizing the reason why genes previously identified as drivers do not appear in the current release (See here). For further inquiries on the reason why a driver is not appearing in this release, please send a request to (bbglab@irbbarcelona.org) and we will try to provide a more detailed explanation.